ILD- IIP and IPF: Diagnosis and treatment approach by Dr. Jyotsna Joshi
M3 India Newsdesk Sep 06, 2019
Dr. Jyotsna Joshi discusses diagnosis and treatment of Interstitial Lung Diseases (ILD), an umbrella term for a group of disorders and how despite there being significant improvement in knowledge of pathogenesis, the aetiology still remains unknown for many cases.

The interstitial lung diseases (ILD) are a group of disorders that predominantly, but not exclusively involve the lung interstitium. The term ILD is imprecise clinical term for a diverse range of diseases that involves inflammation and fibrosis of the alveoli, distal airways, and septal interstitium of the lungs. The lungs are affected in three ways.
- The lung tissue is damaged in some known or unknown way
- This is followed by inflammation of the alveolar wall
- Finally there is fibrosis in the interstitium that results in end stage lung
The stiff lungs cause a restrictive type of functional abnormality and affect gas exchange.
The causes of ILD are many. Despite significant improvement in our knowledge of pathogenesis of ILD, the aetiology of a large number of cases remains unknown and these are called the idiopathic interstitial pneumonias (IIPs). Idiopathic pulmonary fibrosis (IPF) is the commonest example of this group.
ILDs include a wide variety of conditions, individually, relatively uncommon, but collectively found in approximately 50 people per 100,000 population. There are over 200 specific diffuse lung diseases including those characterized by fibrosing and granulomatous histopathology. Only some are of known aetiology.
It has been suggested that exposure to environmental agents is a major contributory factor in the development of diffuse interstitial lung disease. For example, occupational exposure to beryllium can cause a chronic granulomatous disease that is clinically and histologically indistinguishable from sarcoidosis. A disease resembling IPF results from asbestos exposure, and exposure to cobalt may cause interstitial pneumonia and fibrosis. Other agents that are known to produce diffuse lung disease include some therapeutic drugs, such as amiodarone and radiation.
Identified risk factors include exposure to dust, cigarette smoking, gender, age, and race. For example, sarcoidosis occurs predominantly in the 30- to 40-year age range and is more aggressive in patients of Marwari descent in India. In addition, the male to female ratio is 1:1.5. It has been hypothesised that, because not all individuals exposed to a common environment develop ILDs and because apparently trivial exposure to common environmental agents can result in disease, genetics may play a role.
Causes of Interstitial Lung Diseases
- Congenital: Gaucher's disease, Familial dysautonomias
- Acquired (L M NO P Q and I)
- Lymphphoproliferative disorders: LIP
- Metabolic: Uraemia
- Neoplastic: Primary- Bronchiolo-alveolar carcinoma, lymphomas and Secondary- Lymphangitis carcinomatosis
- Occupational: exposure to organic dust (Hypersensitivity pneumonitis)- e.g. Farmer's lung, Bird fancier's lung, and Humidifier's lung, and inorganic dust (Pneumoconiosis)- e.g. Asbestosis, Silicosis, Coal Worker's Pneumoconiosis
- Physiochemical agents: Gases, fumes, drugs, radiation injury, oxygen toxicity
- Q Circulatory: Vasculitides [Wegner's granulomatosis (WG), Churg Strauss syndrome (CSS)], Thromboembolic disorders, Chronic pulmonary oedema, Hemosiderosis
- I: Infective- Miliary TB, interstitial pneumonias (atypical and fungal); Immunological- Pulmonary eosinophilias, Connective tissue disorders, Sarcoidosis, Pulmonary renal syndromes (Good Pasteur's syndrome); Idiopathic- Chronic fibrosing [Idiopathic interstitial fibrosis (IPF), Non-specific interstitial pneumonias (NSIP)], Smoking related [Respiratory bronchiolitis related ILD (RBILD), Diffuse interstitial pneumonias (DIP)], Subacute/Acute (Cryptogenic organising pneumonias (COP), Acute interstitial pneumonia (AIP)]
- Other causes:
- Pulmonary alveolar proteinosis (PAP)
- Neurofibromatosis
- Inflammatory bowel disease (Ulcerative colitis and Crohn's disease)
- Conditions that mimic ILD: (presence of cysts on HRCT)
- Langerhan cell histiocytosis (LCH)
- Tuberous sclerosis
- Lypmhangioleiomyomatosis (LAM)
Diagnosis of ILD
Clinical Features
Dry cough and dyspnoea are the main symptoms of ILD. Symptoms of the associated disorders like RA, scleroderma, sarcoidosis may be present. When the disease is severe and prolonged, symptoms of right heart failure may occur. Clubbing is a feature of IIPs. Pallor due to anemia of chronic inflammation may also be present in most cases. Raynaud's phenomenon, sclerodactyly, and telangectasia are seen with the CTD. Skin, eye, bone and joint involvement, and hepato-splenomegaly are features of sarcoidosis. The findings of clubbing and exercise desaturation on pulse oximetry are characteristic findings on general examination. On respiratory examination bibasilar, end inspiratory crackles or "velcro rales" are noted.
Investigations
- Chest X-ray, pulmonary function tests, and blood tests are important baseline tests.
- High-resolution computerised tomography (HRCT) is an important new diagnostic tool in the evaluation of ILD.
- Lung biopsy, trans-bronchial (TBLB) or open (OLB) may be required for diagnosis and to predict response to therapy in some cases.
- To monitor disease progression and response to therapy symptoms, pulmonary function tests (PFT) and 6 minute walk test (MWT) are used.
Laboratory Tests
The routine laboratory tests are not very useful for diagnosis of ILD except in few cases. Total and differential white cell count (WBC) to diagnose eosinophilia, along with liver and renal function tests should be performed in all cases of ILD. Rheumatoid arthritis (RA) factor and anti-CCP, lupus erythematosus (LE) cells, antinuclear antibody (ANA) anti double stranded DNA (dsDNA), Scl 70 etc. are positive in collagen vascular disorders. Positive cytoplasmic anti-neutrophilic cytoplasmic antibody (C-ANCA) is diagnostic for WG. Serum angiotensin converting enzyme (SACE) is positive in 75% of cases of sarcoidosis but is neither specific nor useful for monitoring response to therapy.
Chest Radiograph
The chest radiograph helps to confirm the clinical diagnosis and in some cases certain radiographic patterns are helpful in establishing a specific diagnosis. The chest radiographs in cases of ILD typically show an "interstitial" pattern, which may be present throughout the lungs or have an upper zone or lower zone predominance. There may be associated alveolar opacities, pleural effusions or hilar and mediastinal lymphadenopathy. In advanced disease enlargement of the right descending pulmonary artery >16 mm suggests pulmonary hypertension and enlarged heart size, cor pulmonale. The chest radiograph is not always very specific and it is sometimes difficult to categorise the opacities as interstitial or alveolar pattern.
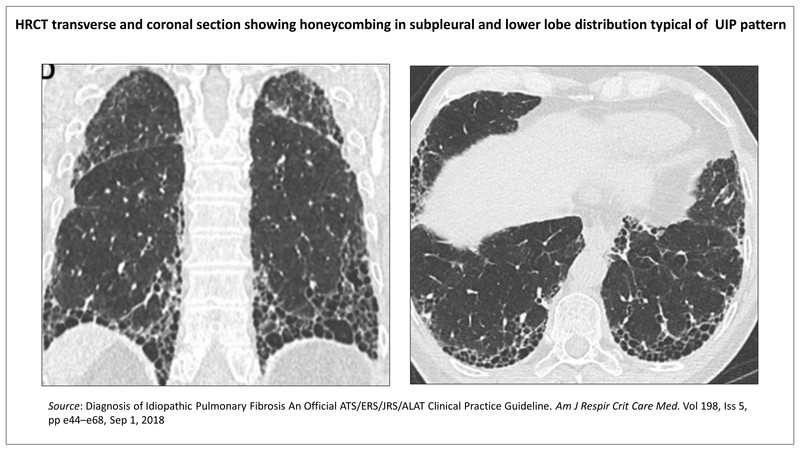
High Resolution Computed Tomography (HRCT)
Computerised tomography (CT) scan of thin section 1-3 mm thickness with narrow field of vision (FOV) for higher magnification and high spatial frequency resolution is called high resolution CT (HRCT). The HRCT is a most useful diagnostic test available for evaluation of ILD. The findings are sufficiently specific in some cases to be diagnostic. HRCT is used to determine whether the images indicate a usual interstitial pneumonia (UIP) pattern, probable UIP, an indeterminate pattern, or an alternate diagnosis. It is also useful to assess the extent of pulmonary, pleural and mediastinal involvement and to guide lung biopsy.
Pulmonary Function Tests and Arterial Blood Gas (ABG) Analysis
Simple spirometry is adequate for initial evaluation and subsequent follow up. Reduced forced vital capacity (FVC) with a normal ratio of forced expiratory volume in 1 second (FEV1) to FVC (FEV1 /FVC) suggests a restrictive abnormality. Diffusion capacity of lung for carbon monooxide (DLCO) is reduced.
Electrocardiography (ECG) and Echocardiography (ECHO)
The findings of p pulmonale, right axis deviation, R /S ratio >1 in V1, ST depression and T wave inversion in V1-V4, and incomplete or complete right bundle branch block on electrocardiography (ECG) identify presence of pulmonary hypertension and cor pulmonale. 2-D ECHO should be performed to confirm pulmonary hypertension and cor pulmonale.
Tuberculin Skin Test (TST)
Tuberculin Skin Testing or Mantoux (MT) testing is important for several reasons in cases of ILD. It may serve as an aid to diagnosis of miliary TB, although in these cases it is often negative due to anergy. Negative MT test supports the diagnosis of sarcoidosis although reports from India suggest that positive test is not incompatible with the diagnosis of sarcoidosis. Positive test is important to initiate isoniazid (INH) prophylaxis along with corticosteroid therapy in cases of ILD. In cases of sarcoidosis a nodule may develop at the site of MT test at 6 to 8 weeks, which can be used for histological confirmation.
Treatment of ILD
The most important aspect of treatment of ILD is early diagnosis and removal of the offending/inciting agent, if identifiable. Specific treatment can be offered in cases of infective aetiology e.g. TB. Early treatment with corticosteroids to reverse the inflammation is vital. In advanced cases symptomatic treatment to provide relief, preventive vaccines to reduce pulmonary infections, oxygen therapy and physical and occupational rehabilitation can be offered to the patients.
There is a desperate need for palliative support for chronic ILDs as survival is sometimes similar to that of advanced malignancy. These patients have very poor survival when admitted to the Intensive Care Unit (ICU) with acute respiratory worsening requiring mechanical ventilation. Similarly, the very small fraction of patients who do survive the ICU stay would require prolonged rehabilitation and could be left with significant morbidity in the form of oxygen dependency, poor mobility, and tracheostomy. Therefore, decision-making process for ventilating or palliating without invasive ventilation.
Efficient patient-centred models of palliative care must take into account religious and cultural differences, as well as variability of resources. Effective implementation of palliative care for ILD will require multidisciplinary participation from clinicians, specialist nurses, psychologists, and social workers. Lung transplantation (single or double) can be performed in end stage lung, and facilities for the same are now available in India, Global hospital Chennai and Mumbai being centres performing lung transplant with good results.
Idiopathic Interstitial Pneumonias (IIPs)
When all known causes of interstitial lung disease have been ruled out, the condition is called idiopathic interstitial pneumonias (IIP). Idiopathic Pulmonary Fibrosis (IPF) is the most common variety of the IIPs. IIP is classified as UIP (Usual Interstitial Pneumonitis) associated with IPF, DIP (Desquamative Interstitial Pneumonitis) and NSIP (Non-Specific Interstitial Pneumonitis). AIP (Acute Interstitial Pneumonia) is a rapid fulminant form of IIP with poor response to therapy and about 90% mortality.
Revised Classification of IIP (ATS/ERS 13)
- Major Idiopathic Interstitial Pneumonia
- Chronic Fibrosing IP: IPF and Fibrotic NSIP
- Smoking –related IP: RBILD and DIP
- Acue/Subacute IP: COP and AIP
- Rare Idiopathic Interstitial Pneumonia
- Idiopathic lymphoid interstitial pneumonia
- Idiopathic pleuroparenchymal fibroelastosis
- Rare histological variants
- Acute fibrosing organising pneumonia(AFOP)
- Airway centered interstitial fibrosis (ACIPF)
- Unclassifiable Idiopathic Interstitial Pneumonias
Idiopathic Pulmonary Fibrosis (IPF)
Diagnosis
It is based upon HRCT and biopsy patterns. Clinically suspect IPF when unexplained symptomatic or asymptomatic patterns of bilateral pulmonary fibrosis on a chest radiograph or chest computed tomography, bibasilar inspiratory crackles, and age greater than 60 years. Middle-aged adults (between 40 and 60 years), especially patients with risks for familial pulmonary fibrosis, can rarely present with the otherwise same clinical scenario as the typical patient older than 60 years.
Investigation
When scan patterns point to probable UIP or indeterminate or alternate diagnosis, patients should undergo bronchoalveolar lavage (showing increased neutrophils and/or absence of lymphocytosis in BAL fluid) and surgical lung biopsy (transbronchial lung biopsy- TBLB and Cryobiopsy are not recommended). Histopathology in UIP/ IPF typically shows dense fibrosis with architectural distortion (i.e., destructive scarring and/or honeycombing), predominant subpleural or paraseptal distribution of fibrosis, patchy involvement of lung by fibrosis, fibroblastic foci and absence of features of an alternate diagnosis. A correct histological classification is of utmost importance based on the fact that prognosis and survival vary largely depending on the subset of IIP.
Treatment protocol
IIPs other than IPF are treated with steroids and immunosuppressives like Azathioprine, Cyclophosphamide. IPF is treated with antifibrotic therapy either Pirfenidone or Nintedanib. Diagnostic certainty is not always necessary before prescribing antifibrotic therapy. A post hoc analysis of the INPULSIS trials found that patients with possible UIP patterns on HRCT scan had similar benefit to those with definite UIP. Although limited data have shown benefit for patients with IPF whose lung function is outside the original inclusion criteria FVC between 50-90% inferences are limited. Given the high cost of the drugs, it is better to observe patients with IPF who have preserved function to gauge the rate of decline prior to initiating nintedanib or pirfenidone.
A recent real-life study showed an increased 3 years survival rate in patients treated with Pirfenidone and a survival benefit of 30% compared to patients treated with no antifibrotic agents. Advantages of dual therapy is uncertain. Treatment of comorbid GERD with antacids is important not only for disease progression but also to treat cough. Benefits of anti reflux surgery is uncertain.
Oxygen therapy should be prescribed for some patients with IPF, particularly those cases with hypoxaemia at rest, presence of pulmonary hypertension and cor pulmonale and exercise induced or nocturnal desaturation. Pneumococcal vaccine and yearly influenza vaccine may help prevent infections.
Pulmonary rehabilitation, palliative care and education programs help to improve the quality of life. Lung transplantation offers hope for people with severe IPF and other advanced ILDs. The Gender Age Physiology (GAP) risk assessment system is a clinical prediction tool that estimates prognosis in patients with IPF (GAP-IPF) and ILDs (ILD-GAP). Multidisciplinary management of ILDs should be done at an experienced centre and the team should comprise of a pulmonologist, rheumatologist, radiologist, pathologist, transplant team and palliative care physician.
Disclaimer- The views and opinions expressed in this article are those of the author's and do not necessarily reflect the official policy or position of M3 India.
-
Exclusive Write-ups & Webinars by KOLs
-
 Daily Quiz by specialty
Daily Quiz by specialty -
 Paid Market Research Surveys
Paid Market Research Surveys -
Case discussions, News & Journals' summaries
